Nurses play a pivotal role in ensuring optimal care.
Takeaways:
- Sickle cell disease is an inherited chronic condition that affects nearly every system in the body.
- Early recognition of complications and comorbidities are essential to limiting morbidity and mortality and improving quality of life.
- Pain is the hallmark symptom of sickle cell disease; nurses are critical to culturally sensitive pain assessment and management.
CNE
1.5 contact Hours
Learning Objectives
- Describe common symptoms and complications of sickle cell disease (SCD).
- Discuss pharmacologic and nonpharmacologic management of SCD.
The author and planners of this CNE activity have disclosed no relevant financial relationships with any commercial companies pertaining to this activity. See the last page of the article to learn how to earn CNE credit.
Expiration: 6/1/24
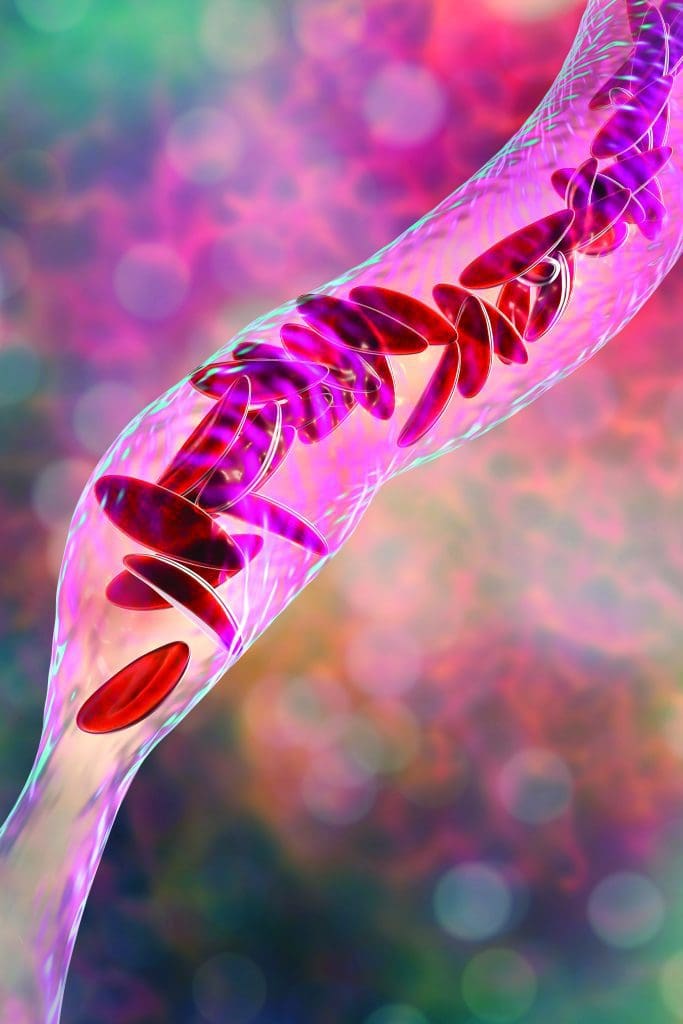
Sickle cell disease (SCD) is an inherited blood disorder (adult hemoglobin [Hb] variants are inherited from both parents) that affects approximately 100,000 people in the United States, 90% of whom are Black. Deoxygenation and polymerization alter the shape of red blood cells (sickling), which leads to blood vessel occlusion and accompanying inflammation, infarction, organ damage, and pain.
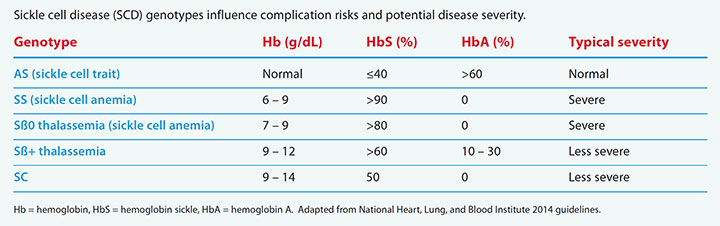
SCD includes multiple genotypes, which depend on the Hb variant contributed by each parent. Typically, individuals with HbSS and HbSß0 thalassemia (also called sickle cell anemia [SCA]) have more severe disease manifestations, although wide variations in severity occur within each genotype. (See SCD genotypes.)
SCD genotypes
Sickle cell disease (SCD) genotypes influence complication risks and potential disease severity.
SCD symptoms typically aren’t seen during the first few months of life because newborns and infants have fetal hemoglobin, which protects against sickling. As the infant becomes older, fetal hemoglobin decreases and symptoms emerge. Historically, those with SCD weren’t expected to survive childhood; however, with the advent of earlier diagnosis (newborns are screened for SCD), improved preventive measures, and better treatments, nearly all children survive into adulthood. The expected life span of someone with SCD varies from 40 to 60 years, depending on genotype and disease severity.
SCD affects nearly every system in the body, so those with the disease have significant healthcare needs and frequently experience comorbidities and premature death. Pain, the most common symptom, frequently begins in early childhood and progresses across the life span. The significant pain associated with SCD negatively impacts psychosocial health and quality of life, which may lead to depression and anxiety.
Psychological health, quality of life, and physical health can be affected by barriers to care such as lack of transportation, inadequate insurance coverage, and challenges balancing other obligations with complex treatment regimens and multiple healthcare appointments. In addition, a systematic review by Bulgin and colleagues found that patients with SCD may experience health-related stigma related to factors such as race, disease status, socioeconomic status, delayed growth and puberty, or having pain that needs to be managed with opioids. (See Wide-ranging effects.)
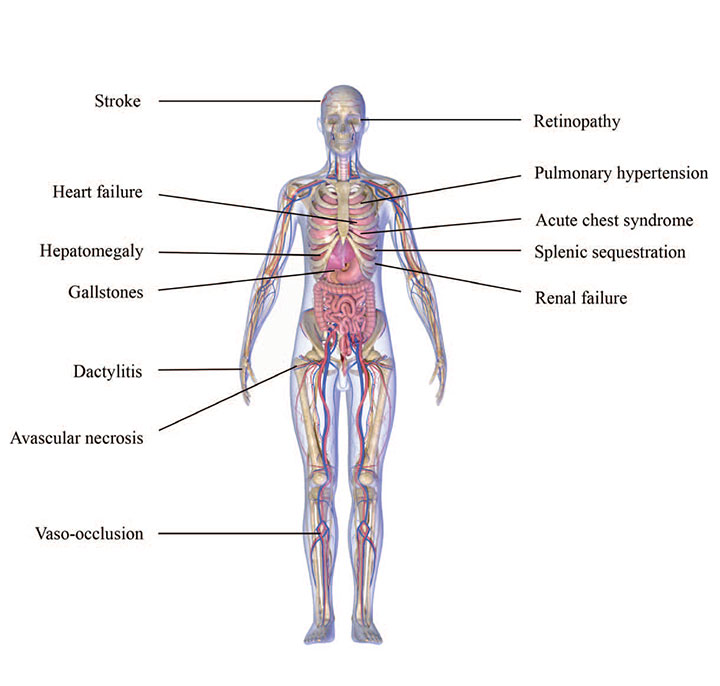
Wide-ranging effects
Sickle cell disease affects most body systems, resulting in comorbidities, frequent hospitalizations, lifelong healthcare needs, and premature death. In addition to the conditions shown below, patients can experience anemia, infection (including sepsis), pressure injuries, priapism, pregnancy complications, and delayed growth and puberty.
Best care practices for SCD include preventive measures to minimize morbidity and mortality, manage symptoms, and recognize severe and life-threatening complications. In addition, nurses must recognize and address psychosocial needs of patients and families and provide education about the disease.
Symptoms and common complications
Pain, fever, sepsis, anemia, splenic sequestration, avascular necrosis, acute chest syndrome, and stroke can occur with SCD.
Pain
Pain is the primary reason individuals with SCD visit the emergency department (ED) or are hospitalized. Pain can be acute, chronic, or acute-on-chronic, and patients may exhibit no objective signs of pain, such as increased blood pressure. Individual experience varies and should be managed by an interprofessional team.
Acute pain crises (vaso-occlusive crises or vaso-occlusive episodes) occur when sickled red blood cells occlude vessels. Pain onset is severe and lasts at least 4 hours with no determinable cause other than blood vessel occlusion. Vaso-occlusive episodes begin as early as 6 months of age and continue throughout life with varying frequency. Individuals with SCA typically experience more frequent episodes. Triggers include dehydration, illness, stress, and temperature changes. These episodes can occur anywhere in the body where vessel occlusion can take place and may occur simultaneously in multiple body parts. The most common locations are the limbs, lower back, abdomen, and chest.
Up to 40% of adolescents and adults with SCD experience chronic pain (lasting longer than 3 months), which may develop over time as the result of the accumulation of complications and organ damage. Those with chronic pain also may have depression and other psychosocial effects such as anxiety and loneliness.
Fever and sepsis
Anyone with SCD is at risk for infection and sepsis, but children, especially young children (<5 years) with SCA, have the highest risk due to hyposplenism and reduced spleen function. This high sepsis risk requires closely monitoring children with fever to quickly identify and treat infection, as well as to assess for other causes of fever such as acute chest syndrome.
Anemia
When the rate of fetal hemoglobin decreases, infants with SCD begin to have anemia that progresses over the next few months or years and persists throughout their lives. Everyone with SCD establishes a baseline state of chronic anemia that typically doesn’t require transfusion. Chronic anemia varies by individual based on several factors, including genotype and other factors. The patient and provider should be aware of the baseline Hb to monitor for acute anemia, which occurs when their baseline drops by 2.0 g/dL or more.
Acute anemia can be caused by multiple factors, including splenic sequestration, delayed transfusion reaction, acute chest syndrome, aplastic crisis, and infection. During acute anemia, providers monitor the reticulocyte count with the complete blood count to assist with determining the etiology and subsequent treatment. Close monitoring can aid early detection of bone marrow dysfunction, which may occur as a result of aplastic crisis.
Splenic sequestration
In young children with SCA, sickled red blood cells trapped in the spleen are common and leads to hyposplenism. Acute splenic sequestration occurs when a large volume of circulatory blood is trapped in the spleen, causing engorgement. It most commonly occurs in children between 1 and 4 years, but depending on the genotype, older children and adults also can experience it.
Signs of splenic sequestration in infants include acute anemia and shock; older children and adults may complain of severe pain. Typically, no cause for the condition can be determined. Because splenic sequestration may recur and result in potentially severe effects, healthcare providers and parents should receive education on palpating the spleen and monitoring for signs and symptoms. Recurrent splenic sequestration frequently requires splenectomy.
Avascular necrosis
Avascular necrosis occurs as a result of blood vessel occlusion in the bones with subsequent ischemia and necrosis. It can manifest in childhood but is most common in adults. The most frequent sites of avascular necrosis are the femoral and humeral heads, but it also can occur in other bones. Signs and symptoms include hip, knee, and shoulder pain. Without treatment, avascular necrosis can lead to permanent damage with chronic pain and impaired mobility. When avascular necrosis is suspected, refer the patient for orthopedic consultation.
Acute chest syndrome
Acute chest syndrome is a medical emergency and a common cause of hospitalization and death in individuals with SCD. It’s seen most frequently in children younger than 10 years, but is more severe in older children and adults. Causes include occlusion and infarction in the lung vessels, inflammation, infection, pulmonary embolism, and bronchospasm. The syndrome can develop slowly or suddenly, and frequently appears similar to pneumonia.
Signs and symptoms include decreased oxygen saturation levels, shortness of breath, fever, and chest pain. In some cases, acute chest syndrome progresses to respiratory failure and multisystem organ failure. Treatment includes incentive spirometry, antibiotics, oxygen, bronchodilators, and transfusion. Children and adults with concurrent SCD and asthma as well as those with prior episodes of acute chest syndrome are at increased risk.
Stroke
Stroke is a severe complication of SCD and a key contributor to disability and death. Patients may have overt (ischemic or hemorrhagic) or silent stroke. Without intervention, children with SCA have an approximately 1 in 10 chance of overt stroke; however, it’s rare in individuals with other genotypes. Overt stroke typically occurs as a result of vessel occlusion or stenosis in the cerebral arteries.
To detect overt stroke risk, children with SCA between ages 2 and 16 years should receive annual transcranial doppler screening. Those with abnormal results should begin chronic red blood cell transfusion therapy for stroke prevention. According to American Society of Hematology guidelines, after 1 year of therapy, individuals interested in stopping transfusions can be evaluated for a switch to hydroxyurea therapy. For children and adults with clinical signs of stroke (overt or silent), providers should immediately administer a blood transfusion (within 2 hours of clinical presentation) before making a definitive diagnosis. After diagnosis, the provider should initiate chronic red blood cell transfusion therapy for secondary stroke prevention.
Silent stroke affects as many as one in three children and one in two adults with SCD and can lead to cognitive impairment and risk for future stroke. Silent strokes frequently occur without noticeable symptoms and commonly aren’t diagnosed without magnetic resonance imaging (MRI). Assess children for developmental delays and both children and adults for cognitive impairment; refer patients and families to a neuropsychologist or psychologist when indicated. In addition, because of the high rates of silent stroke, the American Society of Hematology guidelines recommend that children and adults with SCA receive a one-time screening using MRI to assess for silent strokes.
Priapism
Priapism, which affects over one-third of males with SCD, occurs when the blood vessels in the penis become obstructed and don’t allow normal blood flow, causing persistent erection. Affected individuals should drink fluids, attempt urination, and take a warm bath or shower. Permanent damage can occur over time and may lead to erectile dysfunction and impotence. Priapism lasting longer than 4 hours is a medical emergency.
Other Complications
Other SCD complications include delayed growth and puberty, eye problems (such as retinopathy), gallstones, heart problems (such as heart failure or pulmonary hypertension), kidney problems (such as acute renal failure), leg ulcers, liver problems (such as acute hepatic sequestration), and pregnancy complications (such as preeclampsia).
Treatment
Methods for treating SCD and its symptoms include pharmacologic and nonpharmacologic options, as well as surgery when necessary.
Penicillin prophylaxis
Infants and children with SCA should receive daily oral penicillin for protection against infection and sepsis. Prophylaxis should continue until the child is at least 5 years old but may be continued longer if they’ve had sepsis or splenectomy. Children with other types of SCD may receive daily penicillin if they’ve had a splenectomy. Before ending penicillin prophylaxis, children should receive the complete pneumococcal vaccine series.
Pain management
Management of acute and chronic pain includes pharmacologic and nonpharmacologic therapy.
Pharmacologic therapy. For acute pain management in the acute care setting, assess pain and administer medication within 1 hour of the patient’s arrival. The assessment should include pain characteristics, associated symptoms, location, intensity, and recent analgesic use. Reassess pain every 30 to 60 minutes, and administer additional pain medication as necessary to optimize pain management.
When an individualized pain plan created by the SCD provider is available, treatment should follow it. If a plan isn’t available, base medication selection on findings from the pain assessment, the patient’s home medication therapy, and their experience with effective medications and dosages.
Chronic pain management aims to improve function and quality of life, so opioid therapy should be based on function, goals, and likelihood of sustained benefit over time. For acute and chronic pain, opioids provide the foundation of pain management; however, a multimodal approach that includes non-opioid medications (for example, non-steroidal anti-inflammatory drugs, serotonin and norepinephrine reuptake inhibitors, or gabapentinoids) may optimize pain management.
Nonpharmacologic therapy. Nonpharmacologic approaches are an important part of the pain management plan. Beneficial strategies for acute pain include massage, yoga, virtual reality, and guided relaxation. Individuals with chronic pain may benefit from cognitive behavioral therapy or provider-delivered integrative approaches (such as acupuncture).
Blood transfusion
Acute blood transfusions increase the Hb level and replace abnormal red blood cells with normal cells. Transfusions are administered during complications such as acute stroke, acute chest syndrome, and multi-organ failure. Frequently, individuals with SCD receive preoperative transfusions to avoid postsurgical anesthesia complications, including vaso-occlusive episodes and acute chest syndrome.
Individuals with SCD who’ve had an overt stroke or who are at risk for stroke should receive chronic red blood cell transfusion therapy for at least 1 year to reduce risk of first stroke or repeat stroke. Some will need to continue chronic transfusion therapy indefinitely, but others may be evaluated for transitioning to the maximum tolerated dose of hydroxyurea.
Transfusions may be simple (infusion of donor blood without removal of recipient blood) or exchange (removal of recipient blood before or during donor blood infusion). Exchange transfusion can be manual or via an automated machine.
Chronic red blood cell transfusion therapy typically occurs once every 3 to 4 weeks to maintain HbS levels at a prescribed level (usually less than 30%). Chronic therapy also may be used in individuals who don’t respond to hydroxyurea therapy. Potential complications of chronic transfusion therapy are alloimmunization (immune response after frequent exposure to foreign antigens), infection, and iron overload; risk of iron overload can be reduced with exchange transfusion.
Bone marrow transplant
Bone marrow transplant is the only SCD cure. It requires a genetic match, most frequently a sibling without SCD, and usually is performed in childhood because risks increase in adults. When the donor is a sibling and a good genetic match, about 85% of children have a successful transplant. Potential complications include infection, seizures, graft-versus-host disease, and death. Lifelong medications are required to reduce complication risks.
Medications
Four medications—hydroxyurea, L-glutamine, voxelotor, and crizanlizumab—have been approved to treat SCD and its symptoms. In 1998, the Food and Drug Administration (FDA) approved the oral medication hydroxyurea for adults with SCD. Guidelines now recommend also offering it to children who are 9 months and older. The medication is contraindicated in pregnant and breastfeeding women.
Hydroxyurea increases fetal Hb levels and lowers white blood cell (WBC) and platelet counts, preventing or reducing SCD complications, such as vaso-occlusive episodes, pain, and acute chest syndrome, as well as mortality. It’s recommended for daily use by children and adults with SCA, particularly those with frequent vaso-occlusive episodes, recurrent acute chest syndrome, or severe anemia. It also can be considered in individuals with other genotypes and frequent pain. Adherence is critical to sustain elevated levels of fetal Hb. Low platelet or WBC count side effects may occur; they can be resolved with dose adjustment. WBC, platelet, and reticulocyte counts should be monitored every 2 to 3 months.
In 2017, L-glutamine, an amino acid, was approved by the FDA to treat SCD in those age 5 and older. It’s taken orally twice a day to reduce the number and length of hospitalizations for sickle cell pain and reduce rates of acute chest syndrome.
Voxelotor and crizanlizumab were approved by the FDA in 2019. Voxelotor is an oral medication approved for use in adults and children age 12 and older to prevent sickling and blood vessel occlusion. It decreases red blood cell hemolysis, increasing Hb and decreasing anemia. Crizanlizumab is an I.V. medication approved for use in adults and children age 16 and older to prevent adhesion of red blood cells to vessel walls, reducing occlusion, inflammation, and pain crises.
Nursing implications
Nursing care for patients with SCD focuses on assessing for and identifying complications, advocating for pharmacologic and nonpharmacologic treatment approaches, and individualizing care to meet patient and family needs. (See Key nursing implications.) In addition, nurses are critical to patient and family education, which should include SCD management, how to recognize signs and symptoms of severe complications, and when to notify their provider. AN
Key nursing implications
Nursing best practices when caring for patients with sickle cell disease (SCD) include the following actions.
- Be aware of the stigmatization encountered by individuals with SCD, and provide culturally sensitive care and advocacy, particularly for pain management.
- Conduct a timely and comprehensive pain assessment with prompt pain management.
- Understand that individuals with SCD will likely not have a “typical” pain presentation.
- Recognize that failing to believe that the individual with SCD is in pain contributes to additional suffering.
- Monitor for life-threatening and severe complications, such as acute chest syndrome and stroke.
- Assess for psychosocial complications, and make appropriate referrals.
- Provide patient and family education.
- Promote patient and family self-management.
Shannon Phillips is an associate professor at Medical University of South Carolina College of Nursing in Charleston.
References
Ballas SK, Lieff S, Benjamin LJ, et al. Definitions of the phenotypic manifestations of sickle cell disease. Am J Hematol. 2010;85(1):6-13. doi:10.1002/ajh.21550
Brandow AM, Carroll CP, Creary S, et al. American Society of Hematology 2020 guidelines for sickle cell disease: Management of acute and chronic pain. Blood Adv. 2020;4(12):2656-2701. doi:10.1182/bloodadvances.2020001851
Bulgin D, Tanabe P, Jenerette C. Stigma of sickle cell disease: A systematic review. Issues Ment Health Nurs. 2018;39(8): 675-86. doi:10.1080/01612840.2018.1443530
Chou ST, Alsawas M, Fasano RM, et al. American Society of Hematology 2020 guidelines for sickle cell disease: Transfusion support. Blood Adv. 2020;4(2):327-55. doi:10.1182/bloodadvances.2019001143
DeBaun MR, Jordan LC, King AA, et al. American Society of Hematology 2020 guidelines for sickle cell disease: Prevention, diagnosis, and treatment of cerebrovascular disease in children and adults. Blood Adv. 2020;4(8):1554-88. doi:10.1182/bloodadvances.2019001142
Hassell KL. Population estimates of sickle cell disease in the U.S. Am J Prev Med. 2010;38(4 suppl):S512-21. doi:10.1016/j.amepre.2009.12.022
Jain S, Bakshi N, Krishnamurti L. Acute chest syndrome in children with sickle cell disease. Pediatr Allergy Immunol Pulmonol. 2017;30(4):191-201. doi:10.1089/ped.2017.0814
National Heart, Lung, and Blood Institute. Evidence-Based Management of Sickle Cell Disease: Expert Panel Report. September 2014. www.nhlbi.nih.gov/sites/default/files/media/docs/sickle-cell-disease-report%20020816_0.pdf
National Heart, Lung, and Blood Institute. Sickle cell disease. September 1, 2020. www.nhlbi.nih.gov/health-topics/sickle-cell-disease
Ohene-Frempong K, Weiner SJ, Sleeper LA, et al. Cerebrovascular accidents in sickle cell disease: Rates and risk factors. Blood. 1998;91(1):288-94. doi: 10.1182/blood.V91.1.288
Piel FB, Steinberg MH, Rees DC. Sickle cell disease. N Engl J Med. 2017;376(16): 1561-73. doi:10.1056/NEJMra1510865
Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639-44. doi:10.1056/NEJM199406093302303
Powars DR, Chan LS, Hiti A, Ramicone E, Johnson C. Outcome of sickle cell anemia: A 4-decade observational study of 1056 patients. Medicine. 2005;84(6):363-76. doi:10.1097/01.md.0000189089.45003.52
Smith SK, Johnston J, Rutherford C, Hollowell R, Tanabe P. Identifying social-behavioral health needs of adults with sickle cell disease in the emergency department. J Emerg Nurs. 2017;43(5):444-50. doi:10.1016/j.jen.2017.04.009
Tanabe P, Spratling R, Smith D, Grissom P, Hulihan M. Understanding the complications of sickle cell disease. Am J Nurs. 2019;119(6):26-35. doi:10.1097/01.NAJ.0000559779.40570.2c
U.S. Food & Drug Administration. FDA approved L-glutamine powder for the treatment of sickle cell disease. August 18, 2017. fda.gov/drugs/resources-information-approved-drugs/fda-approved-l-glutamine-powder-treatment-sickle-cell-disease
Vichinsky EP, Neumayr LD, Earles AN, et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group. N Engl J Med. 2000;342(25):1855-65. doi:10.1056/NEJM200006223422502












3 Comments.
Where is the CNE test?
Great article, but it is unclear to me how to take the test for contact hour credit.
Enjoyed the reading, great refresher on SCD. And also update on treatments